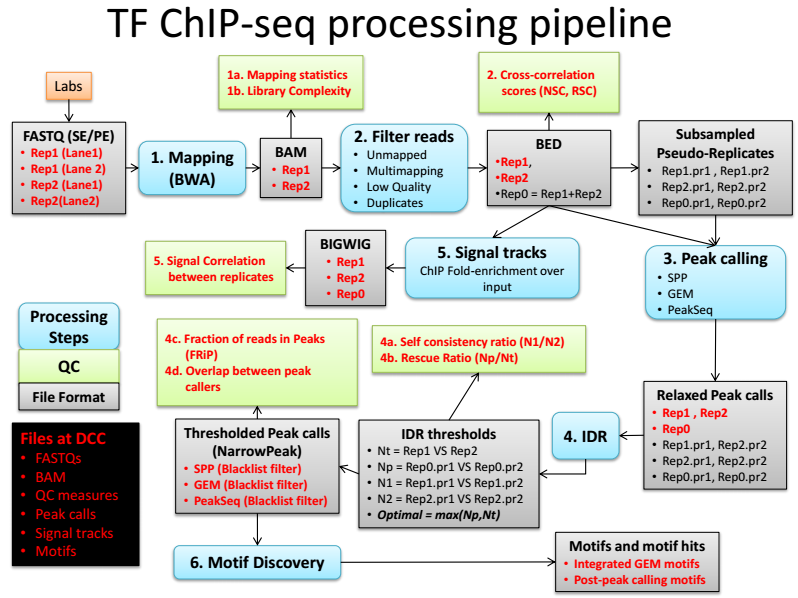
[BI] ChIP-seq data processing using ENCODE pipeline
Link: GitHub - ENCODE3 ChIP-seq pipeline & Google Docs
1. Install miniconda & create conda environment
# Install miniconda
mkdir -p ~/miniconda3
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh -O ~/miniconda3/miniconda.sh
bash ~/miniconda3/miniconda.sh -b -u -p ~/miniconda3
rm -rf ~/miniconda3/miniconda.sh
# Initialize your newly-installed Miniconda
~/miniconda3/bin/conda init bash
~/miniconda3/bin/conda init zsh
# Clone the pipeline.
git clone https://github.com/ENCODE-DCC/chip-seq-pipeline2
Structure of downloaded folder
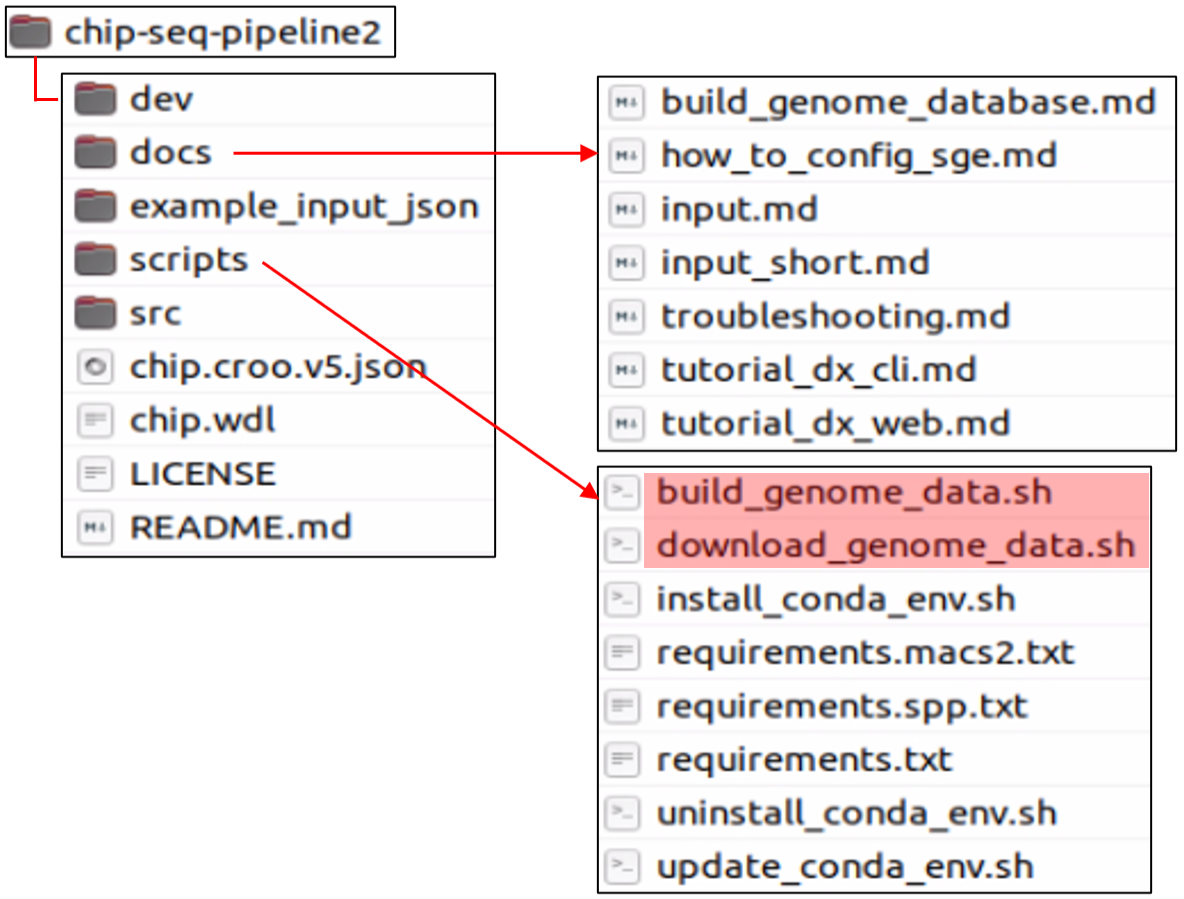
# Install pipeline's Conda environment
bash scripts/uninstall_conda_env.sh # to remove any existing pipeline env
bash scripts/install_conda_env.sh
# Activate created environment
conda activate encd-chip
2. Prepare genome data
Refer the ‘buid_genome_database.md’ file.
# Choose 'GENOME' from hg38, hg19, mm10 and mm9 and
# specify a destination directory.
# ex) bash scripts/build_genome_data.sh [GENOME] [DESTINATION_DIR]
bash scripts/build_genome_data.sh hg38 encode_genome_hg38
3. Pipeline setup
# 1. Install Caper.
pip install caper
# 2. Choose a backend for your system.
caper init local #or slurm, sge, pbs lsf
# 4. Define test input JSON.
INPUT_JSON="https://storage.googleapis.com/encode-pipeline-test-samples/encode-chip-seq-pipeline/ENCSR000DYI_subsampled_chr19_only.json"
# 5. Check if Singularity works on your machine
singularity exec docker://ubuntu:latest echo hello
# 6. Run the test input
caper run chip.wdl -i "${INPUT_JSON}" --singularity --max-concurrent-tasks 1
## --max-concurrent-tasks 1: for computers with limited resources (local)
4. Prepare input JSON file
# Example of input JSON format
{
"chip.title" : "ChIP_A",
"chip.description" : "ChIP_A",
"chip.pipeline_type" : "histone",
"chip.aligner" : "bowtie2",
"chip.align_only" : false,
"chip.true_rep_only" : false,
"chip.genome_tsv" : "encd-genome_hg38/hg38.tsv",
"chip.paired_end" : true,
"chip.ctl_paired_end" : true,
"chip.always_use_pooled_ctl" : true,
"chip.fastqs_rep1_R1" : [ "ChIP_A_1.fq.gz" ],
"chip.fastqs_rep1_R2" : [ "ChIP_A_2.fq.gz" ],
"chip.ctl_fastqs_rep1_R1" : [ "input_1.fq.gz" ],
"chip.ctl_fastqs_rep1_R2" : [ "input_2.fq.gz" ],
}
5. Run the pipeline
for fq in *_1.fq.gz;do
pre=${fq%_1.fq.gz}
caper run chip.wdl -i ${pre}.json --singularity
done

Leave a comment