[BI] ChIP-seq preprocessing witout input control using nf-core pipeline
In ChIP-seq processing, having input control (either fragmented genomic DNA or IgG pulldown) is essential for distinguishing true signal from background noise. However, there are times when I need to analyze ChIP-seq data without the corresponding input samples. Unfortunately, the nf-core ChIP-seq pipeline does not currently support running without input data directly, leaving a gap for users in such situations.
To solve this, I turned to the nf-core GitHub issue page and found that other researchers had encountered the same problem. After exploring the discussions, I discovered a workaround: using the nf-core ATAC-seq pipeline with specific options (--mito_name false --skip_merge_replicates).
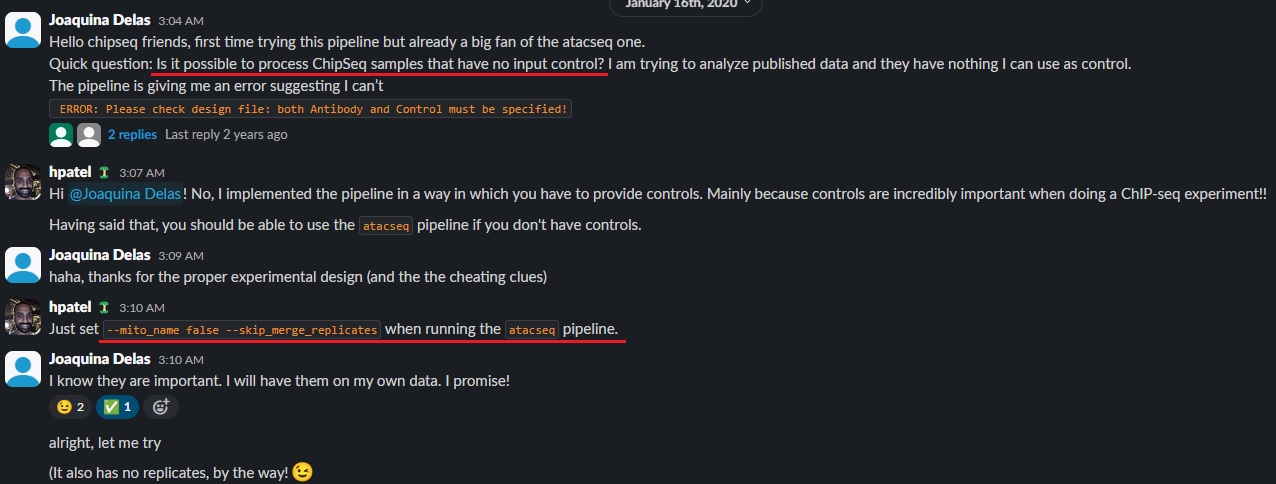
Despite differences in experimental protocols, ChIP-seq, ATAC-seq, and other NGS-based methods like MBD-seq share similar preprocessing steps. These methods all involve fragmenting genomic DNA, and their resulting data often exhibit similar characteristics in the genome. This overlap allows us to repurpose the ATAC-seq pipeline for ChIP-seq processing, even in the absence of input controls.
To test this approach, I ran the nf-core ATAC-seq pipeline with the two specified options on two ChIP-seq datasets. The pipeline produced high-quality outputs and comprehensive QC reports, including MultiQC summaries.
Here’s the exact command I used:
nextflow run nf-core/atacseq -profile singularity \
-r 2.1.2 \
--input samplesheet.csv \
--outdir outdir \
--fasta genome.fa.gz --gtf genome.gtf.gz \
--aligner bowtie2 \
--save_reference \
--macs_gsize 2913022398 \
--mito_name false \
--skip_merge_replicates
--mito_name false: In ATAC-seq, a significant proportion of reads are derived from mitochondrial DNA, which is filtered out. However, this step is unnecessary in ChIP-seq analysis, so setting this flag prevents the removal of these reads.--skip_merge_replicates: Since there may be no replicates to merge, this option ensures the pipeline runs smoothly without trying to combine datasets unnecessarily.
This method offers a practical and efficient solution for ChIP-seq preprocessing without input controls, utilizing existing nf-core tools without requiring custom pipeline development.

Leave a comment