[BI] RNA-seq data processing using nf-core pipeline
Link: nf-core rnaseq pipeline
1. Download the reference genome and genome annotation files
Link: GENCODE
address='https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_45'
wget $address/GRCh38.primary_assembly.genome.fa.gz
wget $address/gencode.v45.primary_assembly.annotation.gtf.gz
2. Prepare a samplesheet
# Move to raw file folder
cd ${path}/00_raw
# Make samplesheet file
echo 'sample,fastq_1,fastq_2,strandedness' > samplesheet.csv
for fq in *_1.fq.gz;do \
pre=${fq%%_1.fq.gz}\
echo "${pre},${pre}_1.fq.gz,${pre}_2.fq.gz,auto" >> samplesheet.csv\
done
Output format
sample,fastq_1,fastq_2,strandedness
BC_0002,BC_0002_1.fq.gz,BC_0002_2.fq.gz,auto
BC_0004,BC_0004_1.fq.gz,BC_0004_2.fq.gz,auto
BC_0005,BC_0005_1.fq.gz,BC_0005_2.fq.gz,auto
BC_0007,BC_0007_1.fq.gz,BC_0007_2.fq.gz,auto
BC_0010,BC_0010_1.fq.gz,BC_0010_2.fq.gz,auto
3. Prepare the alignment index
I run the pipeline for just a sample without the options (–skip_alignment –skip_pseudo_alignment) introduced in Usage for an ‘indexing only’ workflow run, because ‘rsem’ indices are not generated.
# Activate the nf-core environment
conda activate env_nf
# Make samplesheet for a sample
head -2 samplesheet.csv > samplesheet_indexing.csv
# Indexing
fasta="GRCh38.primary_assembly.genome.fa.gz"
gtf="gencode.v45.primary_assembly.annotation.gtf.gz"
outdir="out_index"
nextflow run nf-core/rnaseq -profile singularity \
-r 3.14.0 \
--input samplesheet_indexing.csv \
--outdir $outdir \
--fasta $fasta --gtf $gtf \
--gencode --aligner star_rsem \
--save_reference
## --save_reference: save the indices generated
Output folders structure
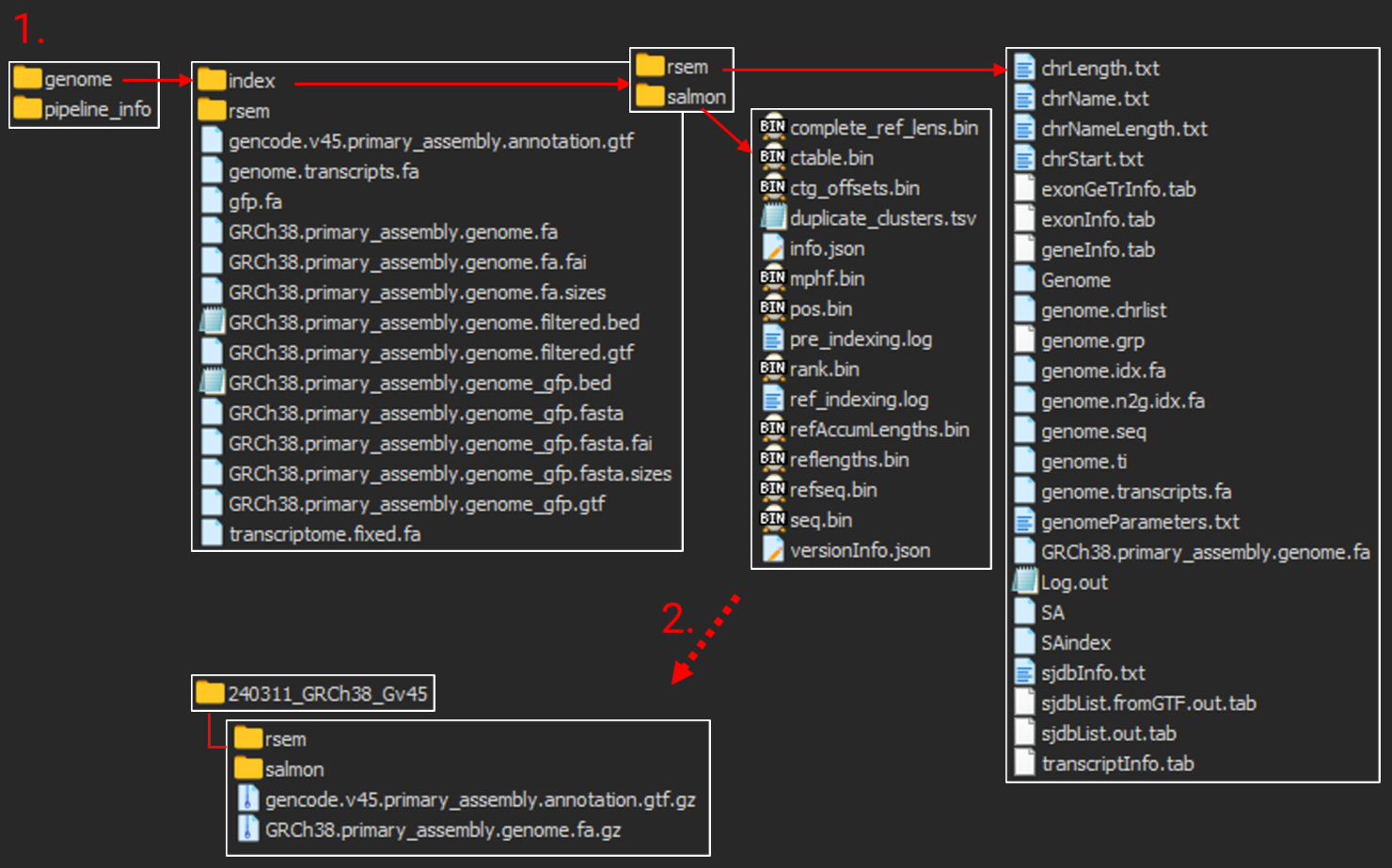
-
Output folders including indices generated
-
A folder restructured for future pipeline runs
4. Run the RNA-seq pipeline
idx="${path}/240311_GRCh38_Gv45/"
outdir="../out_result"
nextflow run nf-core/rnaseq -profile singularity -r 3.14.0 \
--input samplesheet.csv \
--outdir $outdir \
--fasta $idx/*.fa.gz --gtf $idx/*.gtf.gz \
--rsem_index $idx/rsem/ --salmon_index $idx/salmon/ \
--gencode --aligner star_rsem
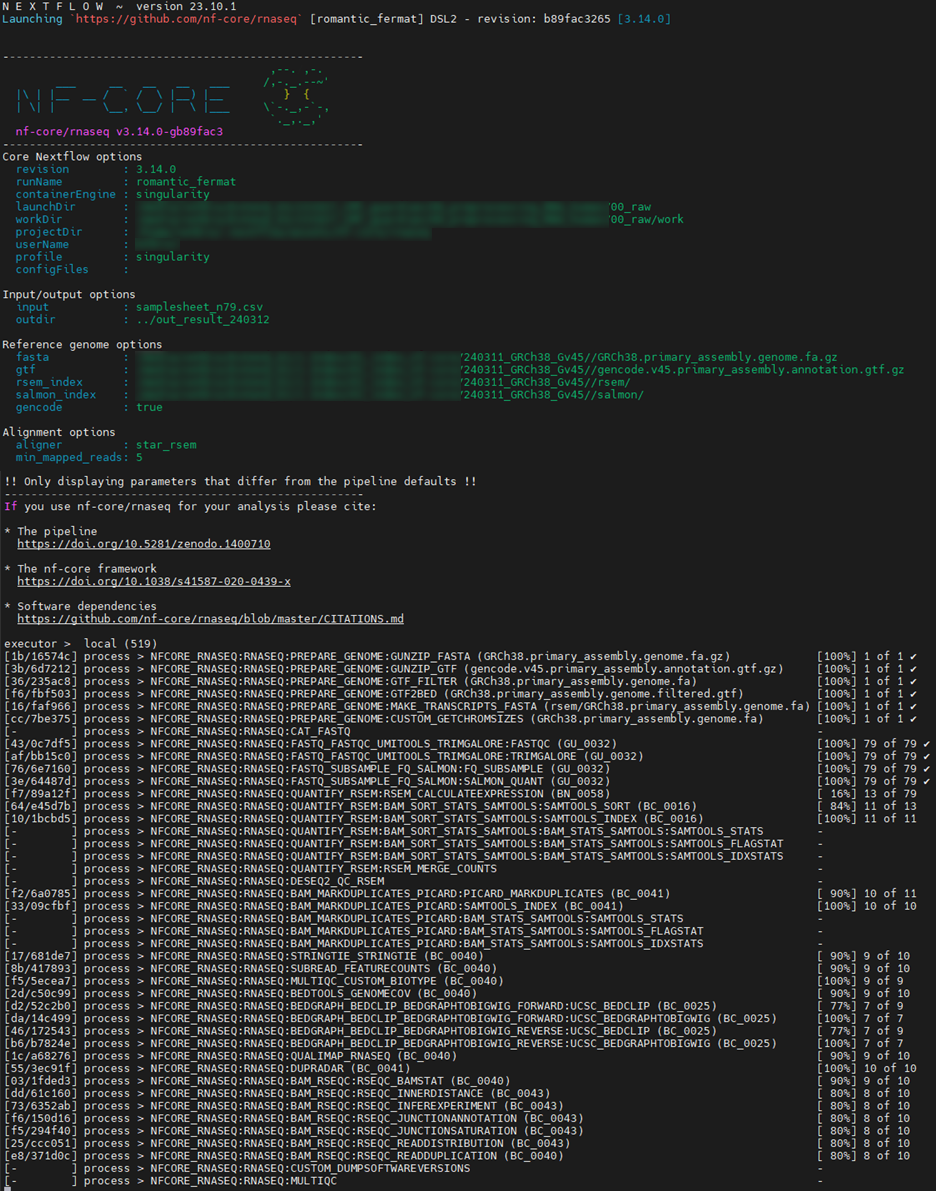
5. Result folder structure
Link: rnaseq: Results

Leave a comment