[BI] Genome Mappability: How to calculate it using Umap
In my previous posts, we explored the concept of problematic regions in genomes, such as the blacklist, and how understanding mappability was important for creating these blacklists. Therefore, in this post, I have outlined the concept of genome mappability and the process of calculating it using the Umap tool. Mappability is a key metric in genomics, especially when working with data from short-read sequencing. While long-read sequencing is becoming increasingly popular, short-read sequencing remains the standard, and the majority of publicly available data is generated from this method.
What is genome mappability?
To grasp the concept of mappability, let’s refer to the abstract from Karimzadeh M. et al. (2018) Umap and Bismap: quantifying genome and methylome mappability. Nucleic Acids Research:
“Every region in a genome assembly has a property called ‘mappability,’ which measures the extent to which it can be uniquely mapped by sequence reads. In regions of lower mappability, estimates of genomic and epigenomic characteristics from sequencing assays are less reliable. These regions have increased susceptibility to spurious mapping from reads from other regions of the genome with sequencing errors or unexpected genetic variation.”
Why use Umap?
While several tools are available for calculating mappability, this post will focus on Umap, a tool capable of calculating both genome and methylome mappability.
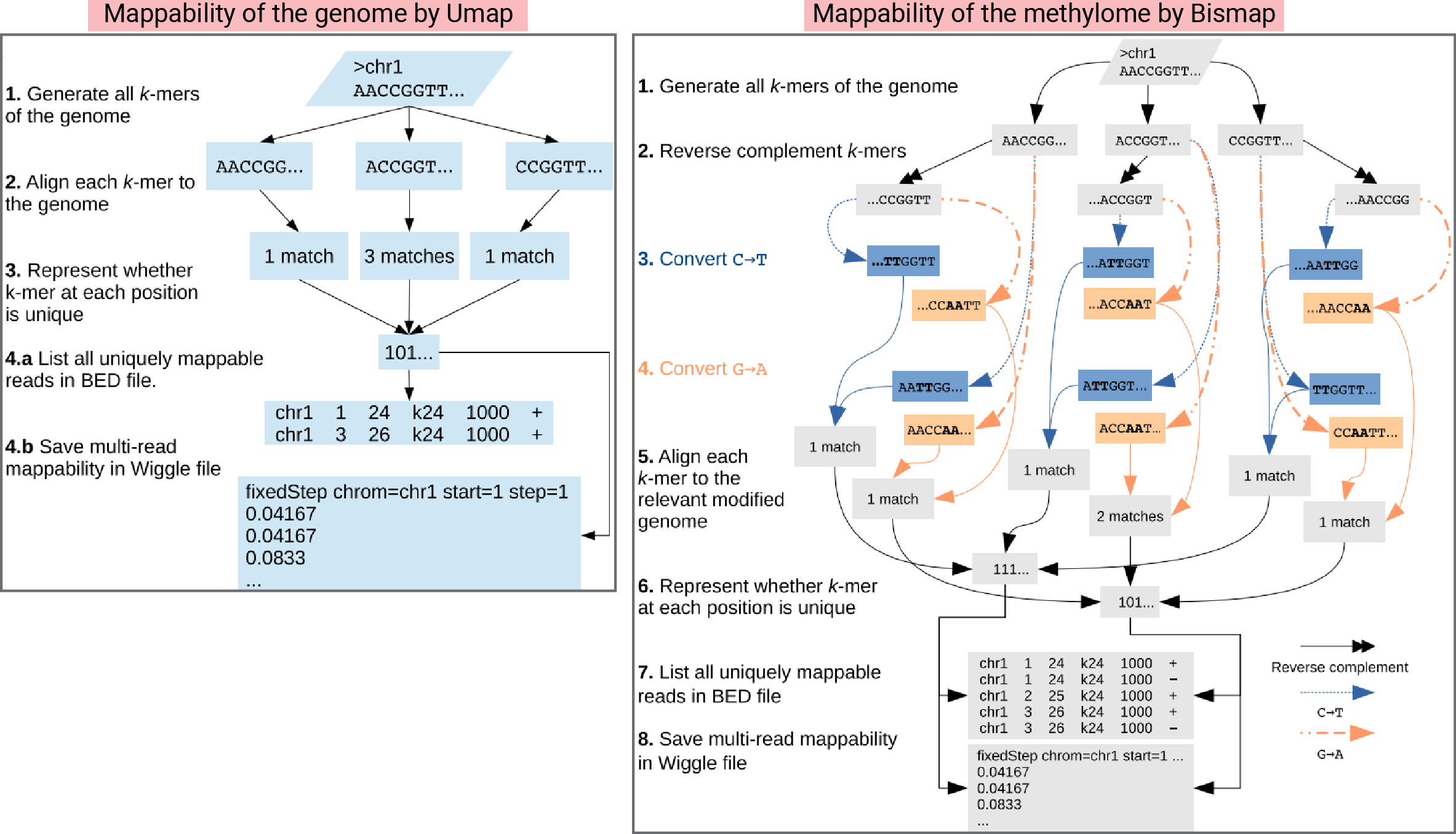
How to use Umap?
1. Setup
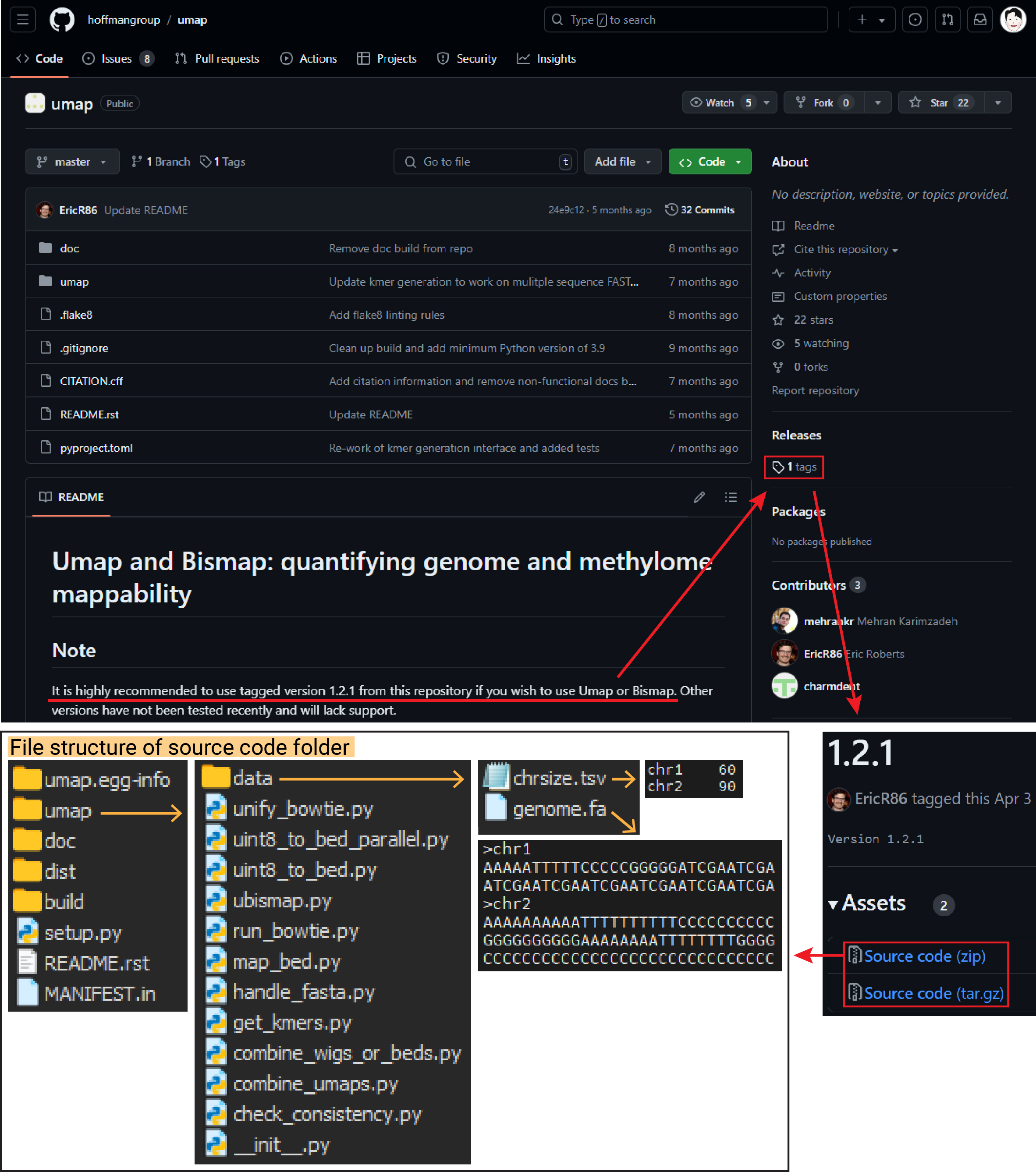
1) Download the source code
Umap’s latest version is v1.2.1, and the source code can be downloaded from the GitHub page (hoffmangroup/umap) through the tags. You can also understand the file structure from the image below.
2) Create separated conda environment
Since Umap is compatible only with Python 2.7, create a separate environment in Conda as follows:
$ conda create --name py2_env python=2.7
$ conda activate py2_env
3) Requirements (tools & python packages)
To ensure Umap functions correctly, install the followings:
$ pip install argparse numpy pandas # Python packages required for Umap
$ pip install gencube # For downloading genome assembly files
$ sudo apt update # Install GNU parallel for multi-threading
$ sudo apt install parallel
$ conda install bioconda::samtools # For genome indexing
$ conda install bioconda::bowtie # For k-mer reads mapping
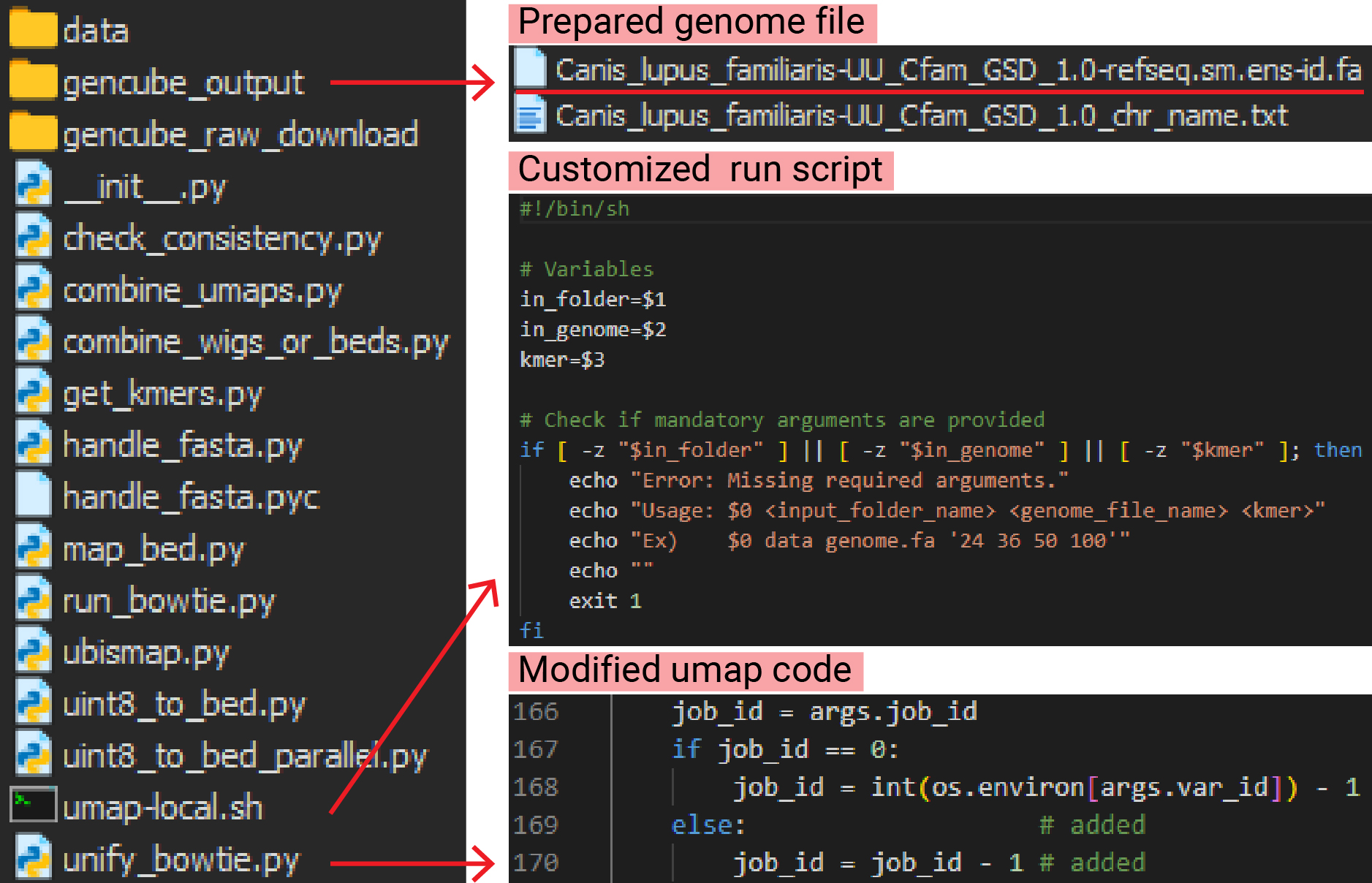
4) Code modification for error resolution
While working with Umap, I encountered an error that required a small code modification. The necessary changes are as follows:
# Original Code
167 if job_id == 0:
168 job_id = int(os.environ[args.var_id]) - 1
# Modified/added Code
169 else: # added
170 job_id = job_id - 1 # added
5) Download customized run script.
The scripts generated by ubismap.py are designed with the assumption that you are using a Sun Grid Engine cluster, which is a type of computing cluster that manages job scheduling and resource allocation using the Sun Grid Engine workload manager. However, if you need to run it on a local workstation, you must modify the scripts.
The author provided guidance on how to modify the script for SLURM, another workload manager similar to Sun Grid Engine but also capable of running on local PCs. Since I preferred a simpler solution, I wrote a script to run using parallel instead. I’ve uploaded the customized script to GitHub (keun-hong/umap-local). Download the file and place it in the Umap folder.
# Clone the repository and modify script permission
$ git clone https://github.com/keun-hong/umap-local.git
$ chmod +x umap-local/umap-local.sh
# Display usage instructions
$ ./umap-local/umap-local.sh
Error: Missing required arguments.
Usage: ./umap-local/umap-local.sh <input_folder_name> <genome_file_name> <kmer>
Ex) ./umap-local/umap-local.sh data genome.fa '24 36 50 100'
2. Run the customized script
1) Prepare genome assembly
Using the Gencube tool, you can easily download the desired genome with a single command. By running the command below to download the dog genome (canFam4), the necessary file for mappability calculation will be prepared, as shown in the below image.
# Download the genome assembly
gencube genome GCF_011100685.1 --download
# Decompress the downloaded genome file
gzip -d gencube_output/*.fa.gz
2) Run the customized script
input_folder_name="gencube_output"
genome_file_name="Canis_lupus_familiaris-UU_Cfam_GSD_1.0-refseq.sm.ens-id.fa"
$ ./umap-local.sh $input_folder_name $genome_file_name '24 36 50 100'
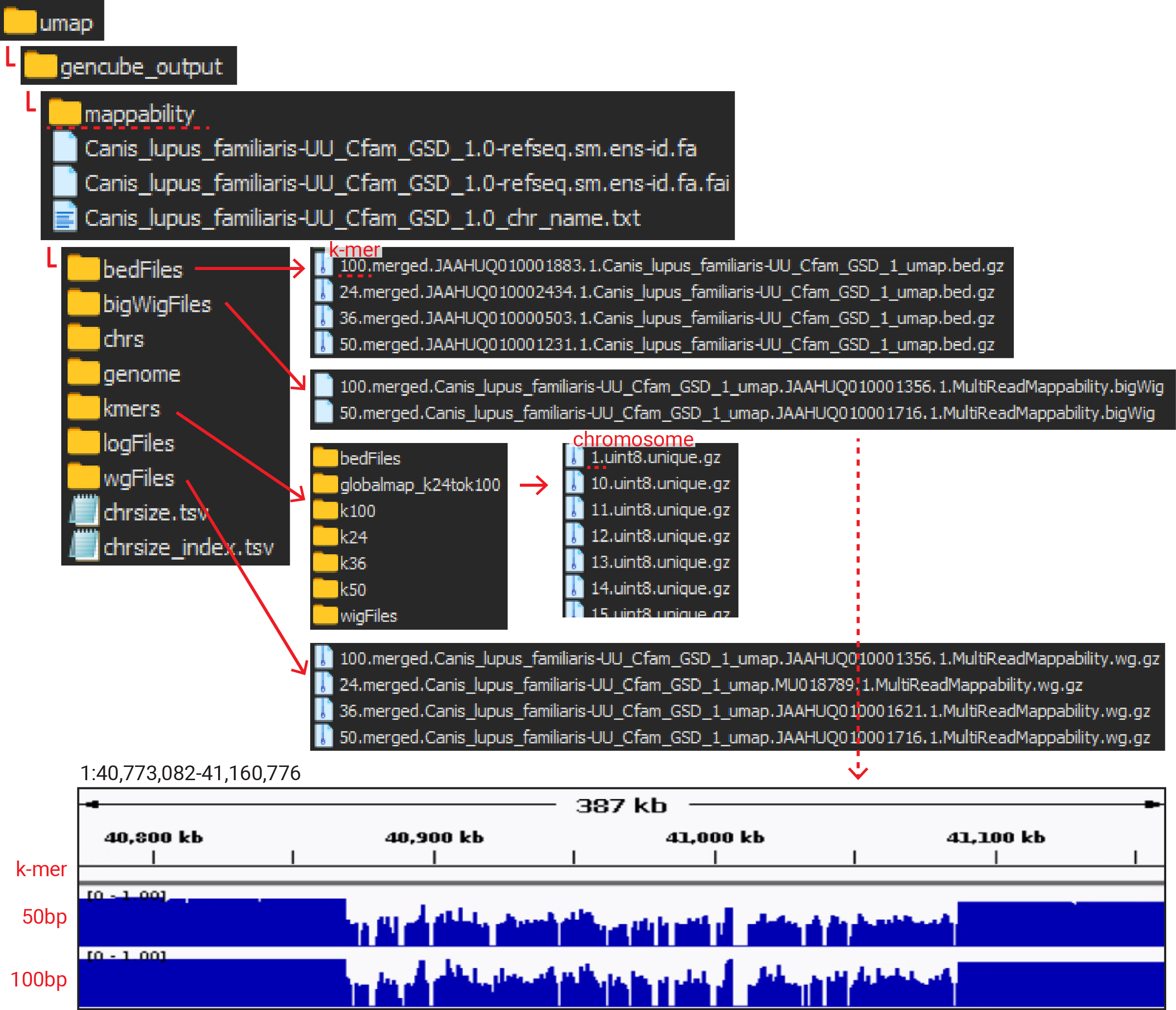
3. Outputs
I used 22 threads, and the mappability calculation for the dog genome (approx. 2.5 Gb) took about 30 hours in total, resulting in the following output.

Leave a comment