[DB] INSDC: How to download NGS raw data
In my previous post, I described how to specifically search for public NGS-based data and fetch metadata from INSDC database. Today I hope to talk with you about how to download the selected raw data.
To download the data, several method or tools as listed below can be used. In this options, I choose the nf-core/fetchngs pipeline due to several reasons.
-
File links searched from SRA Explorer (GitHub)
Why I use nf-core/fetchngs pipeline?
-
Portability & Reproducibility:
nf-core pipelines, like fetchngs, enable consistent and reproducible analyses across various environments, streamlined by containerization (ex. Singularity). -
Scalability:
Fetchngs efficiently manages simultaneous downloads of multiple files, enhancing throughput and expediting data acquisition in large-scale studies. -
Re-entrancy:
Fetchngs supports restarting downloads from failure points, conserving time and computational resources with-resumeoption. -
Connectivity:
Designed for direct interoperability, fetchngs seamlessly connects with nf-core’s data processing pipelines (rnaseq, atacseq, viralrecon & taxprofiler) for integrated analyses.
How to use?
Required input file
To run the pipeline, an input file containing accession numbers per line is required.
vim ids.csv
ERX2744752
ERX2744753
SRX15288999
SRX15289000
How to set-up and run?
# Download Singularity images of fetchngs pipeline
nf-core download fetchngs \
-r 1.2.0 -d -x none -s singularity -u amend
# Confirm the usage
nextflow run nf-core/fetchngs --help
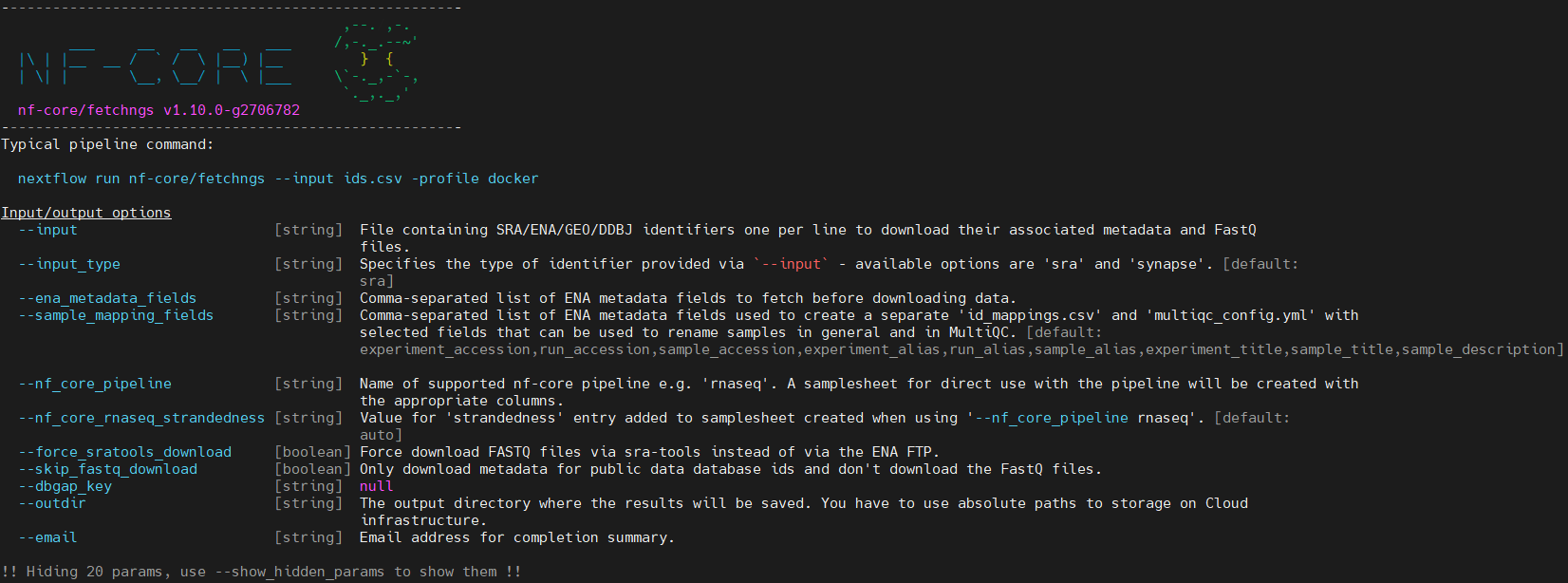
# Run the pipeline
nextflow run nf-core/fetchngs \
-profile singularity \
--input ids.csv \
--outdir 00_raw \
-resume # if error is occured.
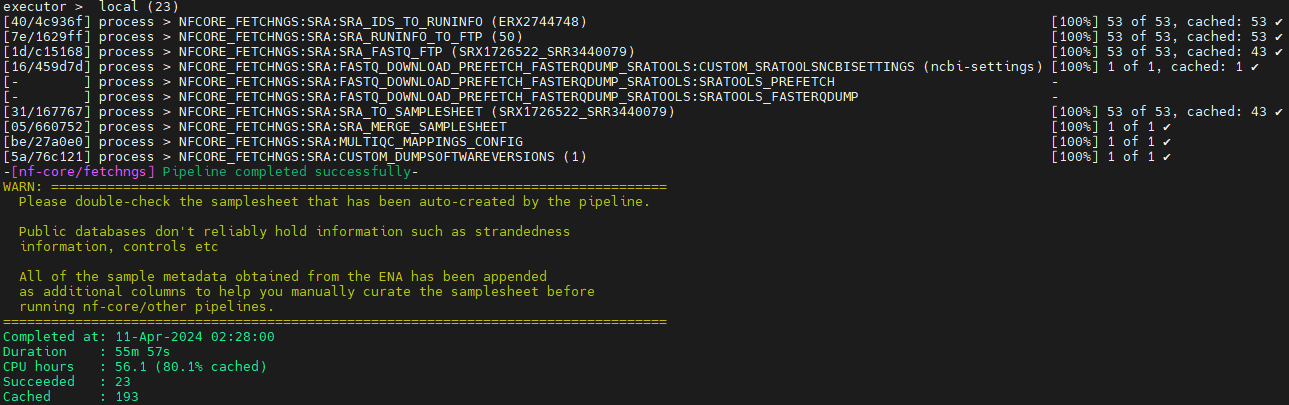
Results
Link: fetchngs: Results

Other - File link searched from SRA Explorer

Reference
2109_Nmeth) Reproducible, scalable, and shareable analysis pipelines with bioinformatics workflow managers
2002_Nbiotech) The nf-core framework for community-curated bioinformatics pipelines

Leave a comment